
Fabry Disease Mouse Models for Preclinical Efficacy Evaluation


Fabry disease (FD) is a rare, progressive, X-linked inherited disorder caused by deficient or absent lysosomal α-galactosidase A (α-GAL) activity, leading to impaired glycosphingolipid metabolism. The prevalence of FD is estimated to range from 1 in 40,000 to 1 in 117,000 in the general population.
The core mechanism of FD is the disruption of globotriaosylceramide (GL3) degradation. Reduced α-GAL activity results in the progressive accumulation of GL3 and its deacylated derivative, lyso-GL3 (globotriaosylsphingosine), within various organs such as the heart, kidneys, and the cerebrovascular system. Due to this direct link to disease pathology, plasma lyso-Gb3 concentration is widely used as a reliable biomarker for evaluating treatment efficacy.
FD is primarily caused by mutations in the GLA gene, which lead to the loss of function and activity of the α-Gal A enzyme. Consequently, GLA gene knockout mice (Gla-KO) have become the preferred model for preclinical evaluation of potential therapies.
To accelerate FD research and help researchers better understand disease mechanisms and develop new treatments, GemPharmatech has developed two GLA gene knockout mouse models:
Gla-KO (Strain No. T012717): Built on a C57BL/6JGpt genetic background.
NCG-Gla-KO (Strain No. T057359): Built on an immunodeficient NCG background.
These models provide critical tools for advancing therapeutic discovery in Fabry disease.
Strain Name | Gla-KO | NCG-Gla-KO |
Description | · Decreased Gla mRNA expression and enzyme activity · Increased Gb3 and Lyso-Gb3 levelsin plasma and multiple tissues | · Decreased Gla mRNA expression and enzyme activity |
Application | · Evaluation of enzyme replacement therapy, molecular chaperones, gene therapy, etc. | · Evaluation of enzyme replacement therapy, molecular chaperones, gene therapy, stem cell therapy, etc. · Suitable for studies requiring immunocompromised host to accept human cells |
Gla-KO
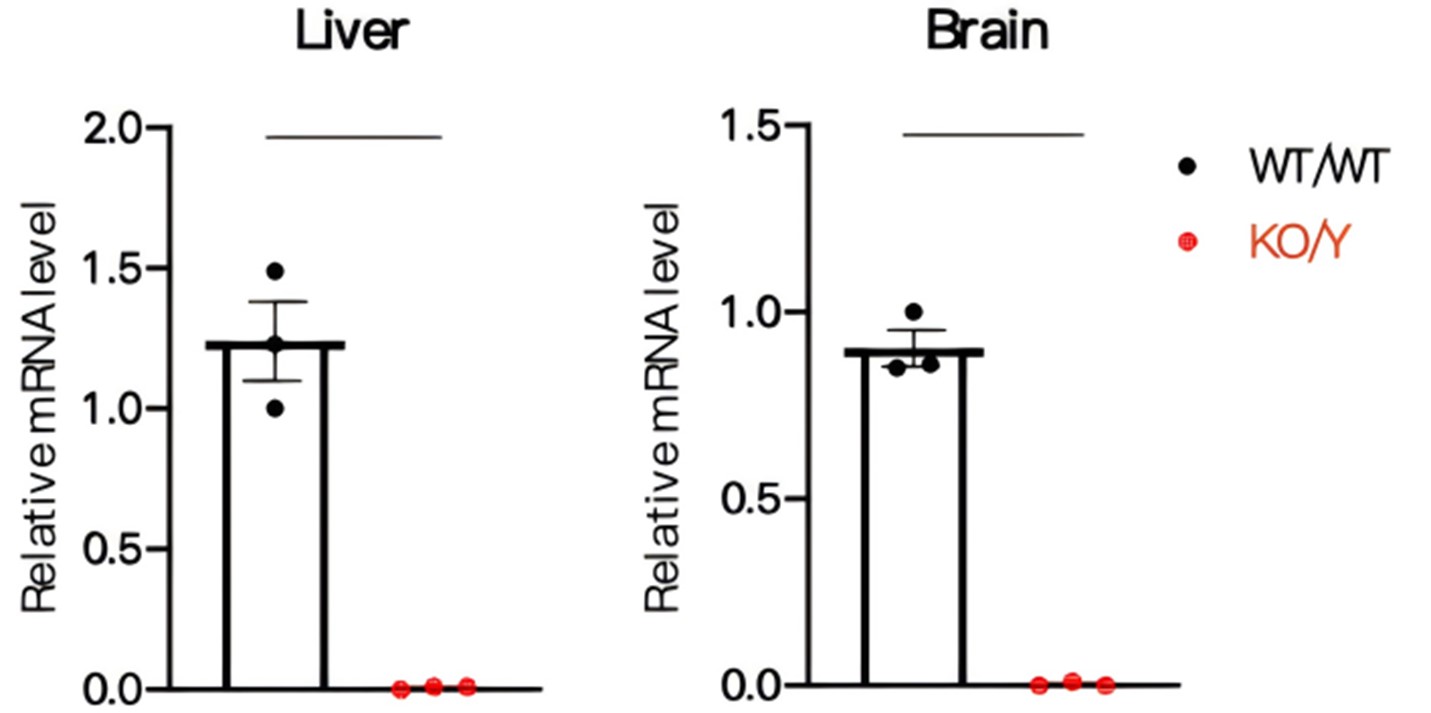
Figure 1 Expression levels of Gla mRNA in Gla-KO mice
Compared with wild-type mice, Gla mRNA levels in the liver and brain of 25-week-old mice hemizygous Gla-KO male mice were significantly reduced. Data were shown as mean±SEM. WT/WT: C57BL/6JGpt wildtype mice, KO/Y: male Gla-KO heterozygous mice.
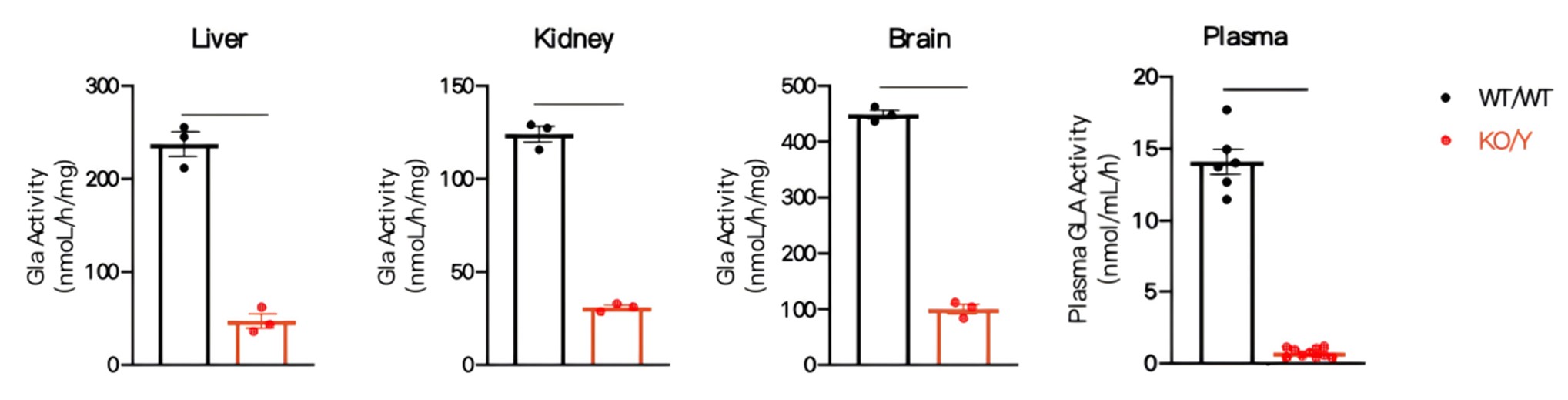
Figure 2 Gla enzyme activity in Gla-KO mice
Compared with wild-type mice, Gla enzyme concentration was markedly decreased in plasma and multiple tissues(including liver, kidney and brain tissues) of hemizygous 19/25-week-old Gla-KO male mice. Data were shown as mean±SEM. WT/WT: C57BL/6JGpt wildtype mice, KO/Y: male Gla-KO heterozygous mice.
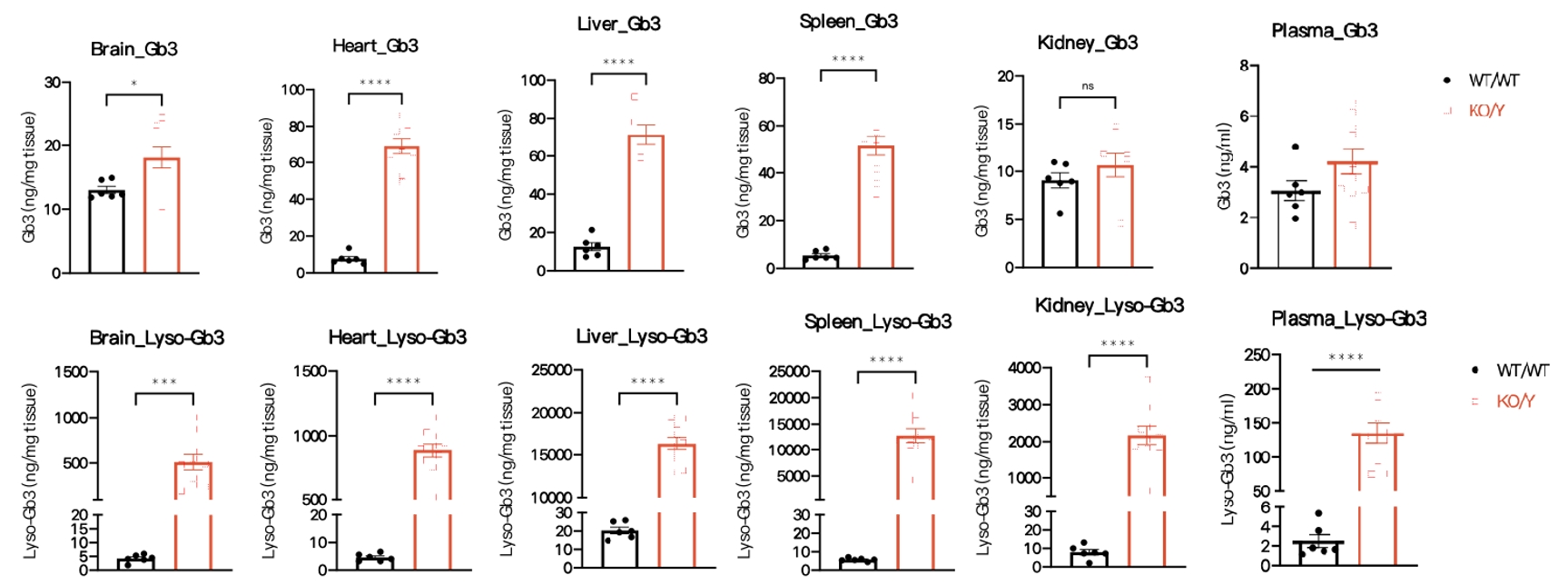
Figure 3 Gb3 and Lyso-Gb3 concentration in Gla-KO mice
Compared with wild-type mice, Gb3 and Lyso-Gb3 protein levels were significantly increased in plasma and multiple tissues(including brain, heart, liver, spleen, kidney tissues) of hemizygous 19/25-week-old Gla-KO male mice. Data were shown as mean±SEM. WT/WT: C57BL/6JGpt wildtype mice, KO/Y: male Gla-KO heterozygous mice.
NCG-Gla-KO
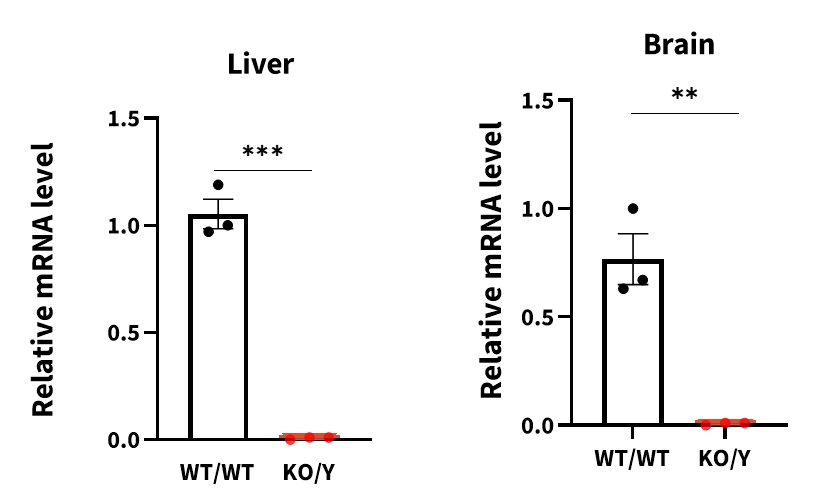
Figure 4 Expression levels of Gla mRNA in NCG-Gla-KO mice
Compared with wild-type mice, Gla mRNA levels in the liver and brain of 25-week-old mice hemizygous NCG-Gla-KO male mice were significantly reduced. Data were shown as mean±SEM. WT/WT: C57BL/6JGpt wildtype mice, KO/Y: male NCG-Gla-KO heterozygous mice.
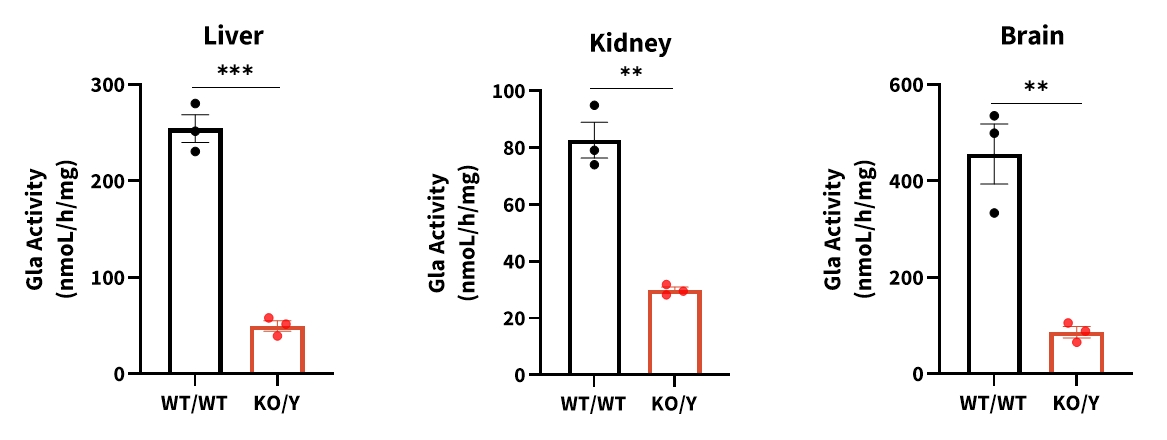
Figure 5 Gla enzyme activity in NCG-Gla-KO mice
Compared to wild-type mice, Gla enzyme activity was significantly decreased in liver, kidney and brain tissues of hemizygous 25-week-old NCG-Gla-KO male mice. Data were shown as mean±SEM. WT/WT: C57BL/6JGpt wildtype mice, KO/Y: male NCG-Gla-KO heterozygous mice.
*Annotation for the samples used in each figure: plasma samples were collected from 19-week-old mice, while tissues were collected from 25-week-old mice.
A Comprehensive Platform for Metabolic Disease Research
Beyond Fabry disease, GemPharmatech supports a broad spectrum of metabolic rare disease research with our comprehensive portfolio of precision models, including:
Disease Type | Strain Name |
Arginase Deficiency | Arg1 CKO |
GM1 Gangliosidosis | |
Hereditary Fructose Intolerance | |
Hyperammonemia | |
Methylmalonic Acidemia | |
Ornithine Transcarbamylase Deficiency (OTCD) | |
Wilson Disease |
Advancing Discovery Through Precision Models
Your next discovery in Fabry disease or related metabolic disorders deserves a foundation of reliable, data-validated science. At GemPharmatech, we provide more than just models—we deliver the clarity and confidence needed to propel your research forward.
Ready to see how the right model can transform your pipeline? Contact us at sales@gempharmatech.com to discuss your project needs.